Tubulointerstitial & microvascular compartment diseases: Clinical evaluation: Dr. NK Hase's Exclusive Masterclass Series Part 3
M3 India Newsdesk Sep 16, 2020
Dr. NK Hase delivers a masterclass on kidney diseases, exclusive in this weekly series for M3 India. In the third part, he writes on diseases of the tubulointerstitial and microvascular compartments. If you have queries, send in your questions at the end of the article. Dr. Hase will address them in a separate article, once this series concludes.
For our comprehensive coverage and latest updates on COVID-19 click here.
Tubulointerstitial compartment
Tubulo-interstitium includes tubules which comprise about 80% of the kidney volume. The interstitium is composed of interstitial cells and a loose, flocculent extracellular material rich in glycosaaminogycans. In the cortex there are mainly glomeruli, interstitial tissue is sparse. In the medulla there is gradual increase in the amount of interstitial tissue from outer medulla to the papillary tip. Renal tubules start from the Bowman’s capsule. It consists of proximal convoluted tubules lies in the cortex and is lined by cuboidal epithelium with brush borders which help to increase the area of absorption. The next loop of Henle extend from proximal tubule to thick ascending limb of loop and then distal convoluted tubule and collecting duct. Finally, the collecting duct opens into minor calyces through the papilla.
This compartment of kidney performs a critical function of selective reabsorption and excretion of glomerular filtrate. It also produces erythropoietin, active vitamin D, rennin and prostaglandin. From 100 ml of protein-free filtrate, 1ml of urine is formed per minute, i.e. 99% of filtrate is reabsorbed by tubules.
The tubulo-interstitium maintains normal homeostasis by tubular transport, reabsorption, and excretion. It is responsible for maintaining electrolyte balance, volume status, osmolality, acid base balance and calcium phosphate balance. It takes part in guconeogenesis and glucose metabolism. It produces active vitamin D3, erythropoietin for maintaining calcium, phosphate and haemoglobin respectively. It also maintains blood pressure by producing rennin, endothelins and prostaglandins.
The tubulointerstitium have high energy demand and are susceptible to injury and insult by various aetiologies like hereditary diseases, ischaemia, drugs, infections, toxins, heavy metals, metabolic disorders, obstructive uropathy, and reflux nephropathy. The tubulointerstitial compartment is also secondarily affected in glomerular and vascular diseases. Long term prognosis depends upon the degree of interstitial fibrosis and tubular atrophy.
Clinical manifestation and presentation of tubulointerstitial involvement
Tubulointerstitial involvement commonly presents as:
- Acute interstitial nephritis: Clinical presentation AKI.
- Chronic interstitial nephritis: Clinical presentation CKD.
- Acute tubular necrosis: Clinical presentation AKI.
- Tubular Syndromes: Due to transporters and channel dysfunction
- These dysfunctions may cause fluid loss and abnormalities in electrolyte and acid-base homeostasis (figure 3)
- Fanconi syndrome: Generalised loss of proximal tubule function (glycosuria without diabetes mellitus, aminoaciduria, phosphaturia, uricosuria and proximal RTA).
- Renal tubular acidosis (RTA): Normal anion gap (hyperchloremic) metabolic acidosis due to failure of renal acid excretion with almost normal renal function.
- Types of RTA:
- Distal RTA (Type 1): inadequate proton secretion in the distal nephrons
- Proximal RTA (Type2): defective bicarbonate reabsorption in the proximal tubule
- Hyperkalemic Type 4 RTA: impaired aldosterone effect with hyperkalemia and acidosis
- Type 3 (mixed) very rare: Carbonic anhydrase II deficiency.
- Defective urine concentrating ability of tubules: Usually manifests as polyuria, nocturia and isothenuria.
Figure 3: Tubular syndromes

Syndromes of tubulointerstitial injury
1. Acute tubulointerstitial nephritis (ATIN)
Clinical presentation: Abrupt onset AKI, occurs within days of exposure to an offending drug or several months of use of NSAIDs. It may be oliguric or nonoliguric. The patient experiences fatigue,weight loss, anorexia, fever, skin rash, arthralgia, myalgia, flank pain, and haematuria. Clinical features inciting disease, infection, or systemic disease include jaundice, bleeding, subconjuctival haemorrhage in leptospirosis hanta virus infection.
Lab findings:
- Anaemia, eosinophilia (25-40%)
- Sterile pyuria (50%)
- WBC cast ± microhaematuria
- Proteinuria usually absent or mild (less than 1 g/d)
- Protein:creatinine ratio > albumin:creatinine ratio (PCR>ACR)
- Predominantly low mol.wt.proteinuria like beta2 microglobin or alpha-1 microglobin (for diagnosis and monitoring activity)
- Eosinphiluria (low positive predictive value)
- Elevated IgE
- Renal imaging shows normal size or enlarged kidneys on USG
- Gallium isotope scan differentiates from ATN
- Definitive diagnosis by kidney biopsy reveals edematous interstitium with lymphocyte mostly T cells, eosinophils, plasma cells and macrophages, occasionally granuloma formation; glomeruli are usually normal
Aetiology:
- Drug (70%) hypersensitivity reactions (penicillins, sulfonamides, NSAID, rifampin, ciprofloxacin, allopurinol, phenytoin, furosemide, cimetidine, proton pump inhibitors)
- Infections (15%): Bacterial (Salmonella, E.coli, Brucella, Legionella, Leptospira interogans), viral (HIV, CMV, BKV, EBV, HSV, hantaviruses), and fungal (histoplasmosis), and parasitic (toxoplamosis)
- Systemic diseases: Light chain cast nephropathy, Sjogrens syndrome, SLE, tubulointerstitial nephritis and uveittis, sarcoidosis, GPA vasculitis
2. Acute tubular necrosis (ATN)
Clinical presentation: It is the commonest cause of intrinsic acute kidney injury. It may be oliguric or non-oliguric (40%). H/o suggestive of volume depletion like gastroenteritis, haemorhage, hypotension, polytrauma, recent surgery, Sepsis. Cardiac failure, h/o nephrotoxic drug intake.
Lab findings:
- Urine- Protein trace or absent, characteristic finding of muddy brown, granular cast, renal tubular epithelial cell and epithelial cast.
- Fractional excretion of sodium >1%, urinary sodium >40 meq/l, fractional excretion of urea more than 50 (positive predictive value 98%, sensitivity 85%, specificity 92% for ATN), increased BUN, serum creatinine.
- Metabolic acidosis, ifosfamide nephrotoxicity usually presents as a Fanconi syndrome (proximal tubule dysfunction), with significant hypokalemia. Foscarnet nephrotoxicity is commonly associated with hypocalcaemia. Pentamidine nephrotoxicity is frequently associated with hypomagnesaemia and hyperkalemia.
Aetiology:
- Ischaemic tubular necrosis. Prolonged hypotension due to hypovolemia, haemorrhagic, septic, or cardiogenic shock.
- Toxin-induced radiocontrast, rabdomyolysis, intravscular haemolysis, tumour lysis, ethylene glycol, methanol intoxication, snake envenovemation.
- Drug induced: Aminoglycosides, cisplatin, amphotericin B, acyclovir, indinavir crystal induced.
3. Chronic tubulointerstitial nephritis (CTID)
Clinical presentation:
- Insidious slow onset
- Slow, progressive CKD
- Nocturia, polyuria
- Normal BP or mild HT
- No oedema, usually late
- Papillary necrosis with analgesic nephropathy may cause flank pain and haematuria
- Patient may be asymptomatic; detected on screening
Lab findings:
- Anaemia, early in the course of disease due to destruction of erythropoietin producing cells.
- Urine protein may be absent or present in traces (usually less than 1 g)
- PCR>ACR
- Low molecular weight proteinuria-immunoglobulins, beta 2 and alpha 1 microglobulins
- Elevations of serum creatinine
- Early renal tubular dysfunction: Proximal RTA Fanconi syndrome, or dista RTA, hyperkalemia type 4 RTA
Aetiology:
- Drugs: Analgesic, NSAIDs, lithium, cyclosporin, tacrolims
- Heavymetals: Lead, cadmium, mercury
- Reflux nephropathy, obstructive nephropathy, neprolithiasis
- Immunologic diseases: Sjorens syndrome, SLE, sarcoidosis, chronic allograft nephropathy
- Metabolic diseases: Hypercalcemia, hyperuricemia, hyperoxaluria, chronic hypokalemia
- Genetic diseases: Autosomal dominant tubulointerstitial disease Alport disease
- CKD: Mesoamerican nephropathy, Balkan endemic nephropathy, Aristolochia nephropathy (Chinese herb)
Syndromes of renal tubular acidosis
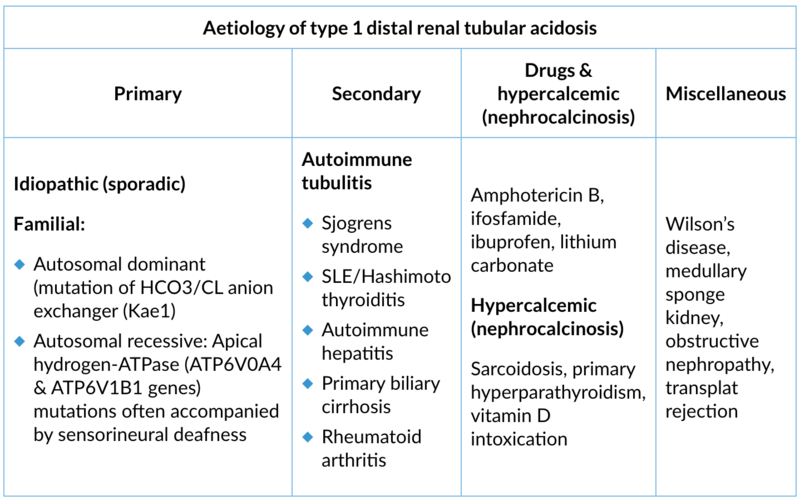
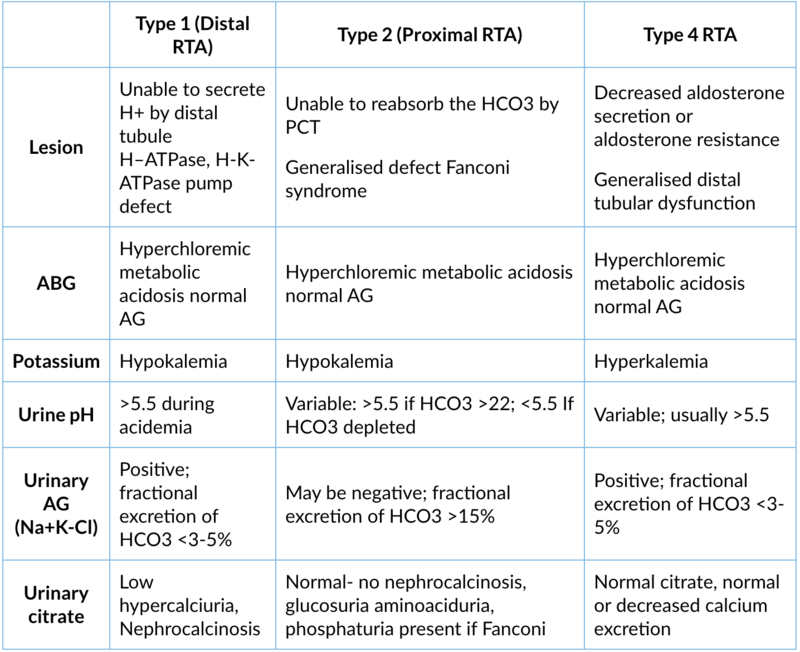
1. Type 1 (distal RTA)
Site of lesion: α-intercalated cells of the distal tubule, H-ATPase, H-K-ATPase pump defect
Type of lesion: Unable to secrete H+ (apical) → ↓ intracellular production of HCO3- → ↓ HCO3-/Cl- exchanger activity (basolateral) → metabolic acidosis
Clinical presentation:
- Polyuria, polydipsia, dehydration
- Growth retardation in children, failure to thrive, rickets in children, osteomalacia in adults
- Muscle weakness, paralysis, hyporeflexia
- ECG suggestive of hypokalemia U wave, flattening of ST segment
- Nephrocalcinosis
Lab findings
- Normal anion gap hyperchloremic metabolic acidosis
- Hypokalemia
- Normal serum creatinine
- Urine pH >5.5
- Urinary anion gap positive
- Decreased ammonia excretion
- Decreased urinary citrate
- Hypercalciuria
- Nephrocalcinosis
- Fractional excretion of HCO3 <3%
- Incomplete distal acid load test: Oral administration of ammonium chloride there is failure to decrease urine PH below 5.3 suggesting acidification defect
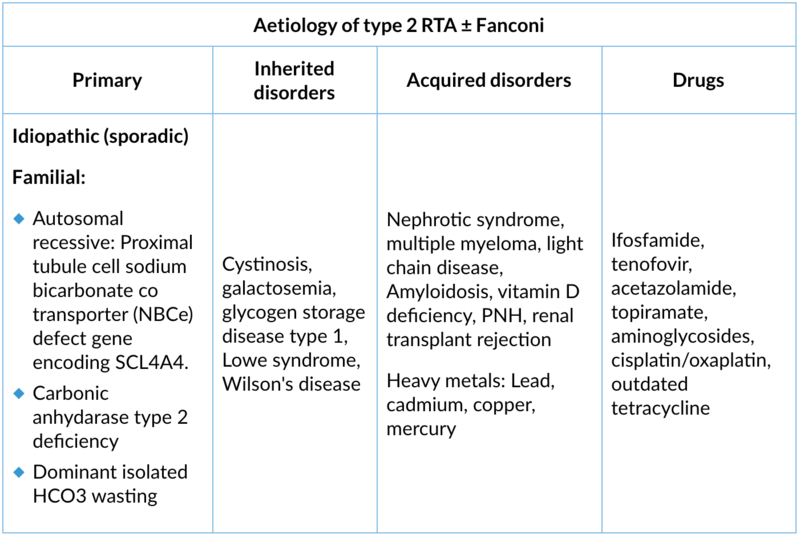
2. Type 2 (proximal) renal tubular acidosis
Site of lesion: Proximal tubule unable to reabsorb the HCO3. It can be isolated HCO3 defect or generalised defect of PCT called Fanconi syndrome
Type of lesion: Only HCO3- reabsorption is defect in isolated type 2 RTA. Impaired reabsorption of HCO3- and other compounds (e.g., potassium, glucose, phosphate, and amino acid reabsorption) in the PCT are called as Fanconi syndrome
Clinical presentation:
- Polyuria, polydipsia
- Short stature
- Growth retardation in children
- Vitamin D resistant rickets
- Muscle weakness, hyporeflexia paralysis because of hypokalemia
- Symptoms are severe in Fanconi syndrome .
Lab findings:
- Normal anion gap hyperchloremic metabolic acidosis
- Hypokalemia
- Normal creatinine
- Urinary pH variable if serum HCO3- is higher than its reabsorption threshold: pH ≥5.5; if serum HCO3- is depleted: pH <5.5
- Urinary AG may be negative
- Fractional excretion of HCO3 >15%
- If Fanconi: Hypouricemia, glucosuria (normal or low blood glucose)
- Phosphaturia
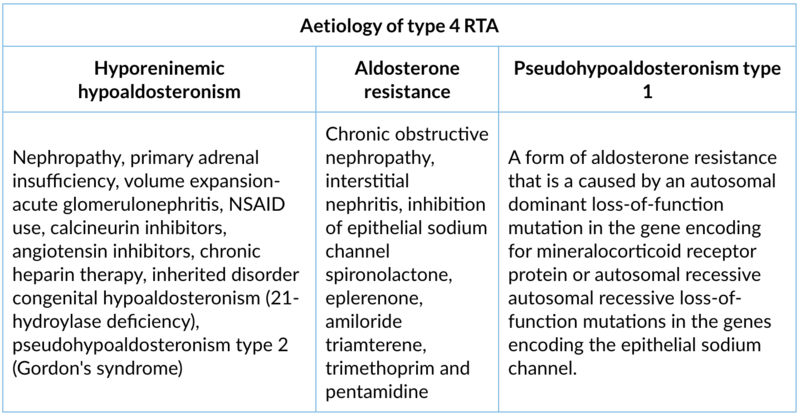
3. Type 4 RTA (hyperkalaemic)
Site of lesion: Generalised distal tubule dysfunction. Decreased aldosterone secretion or aldosterone resistance.
Type of lesion: Aldosterone deficiency and/or resistance cause hyperkalemia and metabolic acidosis which inhibits ammonia synthesis in the proximal convoluted tubule and decreases urinary ammonium excretion.
Clinical presentation: Polyuria, polydipsia, impaired growth in children, muscle weakness, ECG changes of hyperkalemia (tall T waves, prolonged PR interval, widening of QRS complex, sine rhythm and asystole). Features of underlying conditions.
Lab findings:
- Normal anion gap metabolic acidosis
- Hyperkalemia
- Urine pH variable usually >5.3
- Urinary anion gap positive
- Normal or decreased calcium excretion
- No nephrocalcinosis
Comparisons of different RTA
Other tubular syndromes
1. Mixed renal tubular acidosis (type 3)
Rare occurrence combination of Type1 and 2 RTA
Site of lesion: Both proximal and distal tubule, impaired hydrogen excretion by DCT and failure to reabsorb HCO3 by proximal tubule.
Underlying cause: Congenital carbonic anhydrase type 2 deficiency.
Clinical presentation: Osteopetrosis, cerebral calcifications, mental retardation (Guibaud-Vainsel syndrome) autosomal recessive onset in infancy or neonates.
Lab findings:
- Hyperchloremic metabolic acidosis normal AG, hypokalemia, hypocalcemia
- Positive urinary AG, Urine pH >5.3, hyercalciuria, low urinary citrate, increased generalised bone density called as osteopetrosis
Therapy: Alkali therapy, sodium citrate or potassium citrate (Shohl solution)
2. X-linked hypophosphatemic rickets
Site of lesion: Proximal tubule, loss of function mutation in PHEX gene (phosphate regulating endopeptidase on the X chromosome). This encodes enzyme which degrades FGF23. FGF23 inhibits the phosphate reabsorption by sodium/phosphate cotransporter in PCT. Mutation in the PHEX gene leads to increased levels of fibroblast growth factor 23 (FGF23) → indirect inhibition of the sodium-phosphate cotransporter in the proximal renal → impaired reabsorption of phosphate → chronic hypophosphatemia→ vitamin D-resistant rickets/osteomalacia.
Clinical features: Age of onset in childhood (less than 3 years), growth retardation, rickets, bowing of legs, osteomalacia, bone pains, enthesopathy (calcification tendons ligaments and joint capsules, dental pain, dental abscesses, deafness, osteoarthritis and spinal canal stenosis in adults.
Laboratory abnormality: Hypophosphatemia, normal serum calcium, normal to high PTH, normal plasma 25-hydroxy D level, normal or slightly low levels of 1,25-dihydroxyvitamin D, normal serum bicarbonate, urinary pH, nephrocalcinosis, bone changes of rickets/osteomalacia, urinary TmP/GFR is increased suggesting phosphate wasting.
Tubulopathies metabolic alkalosis with hypokalemia
1. Bartter syndrome
Site of lesion: Na+-K+-2Cl- cotransporter in the thick ascending loop of Henle.
Pathophysiology:
- Inactivation results in loss of Cl-, Na+, and K+ in urine (as seen in chronic loop diuretic use)
- Failure to reabsorb Na+ leads to natriuresis (salt and water loss) and volume depletion, which activates the renin angiotensin aldosterone system
- Elevated aldosterone levels leads to increased K+ and H+ excretion and subsequent hypokalemia and metabolic alkalosis
- Decreased paracellular reabsorption of calcium results in hypercalciuria, hypocalcemia and nephrocalcinosis and stones
Types of Bartter and their phenotypes
- Type 1 (SLC12A1 encodes NKCC2) and Type 2 (KCNJ1 encodes ROMK) may present antenatally with polyhydramnios; children are prone to dehydration and growth retardation. Autosomal recessive, polyuria, hypokalemia, metabolic alkalosis, nephrocalcinosis.
- Type 3 (CLCNKB encodes one of the basolateral chloride channels ClC-kb) is the most common form; the phenotype is often mild. The phenotype can be similar to Gitelman. Autosomal recessive, polyhydramnios infrequent, 0-5years onset, polyuria, hypochloremia, metabolic alkalosis, hypokalemia, hypomagnesaemia, variable clinical course, nephrocalcinosis unusual.
- Type 4 is caused by genetic inactivation of both basolateral chloride channels, usually by mutations of the chaperone barttin (BSND); it also causes sensorineural deafness. Antenatal presentation, with polyhydramnios, polyuria, hypochloremia, metabolic alkalosis, hypokalemia, hypomagnesemia, nephrocalcinosis is unusual.
- Type 5 has been assigned to either a Bartter-like syndrome caused by gain-of-function mutations of the calcium sensing receptor (CaSR) or X-linked polyhydramnios and transient infantile salt-wasting (MAGED2).autosomal dominant, polyhydramnios not present, Bartter like hypocalcemia, hypochloremic metabolic alkalosis, hypokalemia, nephrocalcinosis, hypoparathyroidism.
2. Gitelman syndrome
Site of lesion: Distal convoluted tubule- mutation of SLC12A3 gene on chromosome 16p leads impaired function of the thiazide-sensitive sodium-chloride cotransporter.
Pathophysiology: Impaired function of the thiazide-sensitive sodium-chloride cotransporter in the distal convoluted tubule leads to impaired sodium chloride reabsorption resulting in sodium and chloride loss and volume depletion. Mild RAS activation- this results in hypochloremia, hypokalemia, metabolic alkalosis.
Clinical presentation: Age of symptom onset is ≥ 6 years, diagnosis is usually made in adolescence or adulthood, muscle cramps, weakness, fatigue, polyuria dehydration, tetany, normal or low blood pressure.
Laboratory finding: Metabolic alkalosis, hypokalemia, hypomagnesemia, hypercalcemia with hypocalciuria.
3. Liddle syndrome
Site of lesion: Structural alteration in the β and γ subunits of the epithelial sodium channel (ENaC) in the collecting duct due to autosomal dominant ENaC gain of-function mutations.
Pathophysiology: There is structural alteration in the ENaC subunits which leads to failure of subunit to bind with intracellular ubiquitin protein ligase (Nedd4). There is decreased degradation of ENaC channel by ubiquitin proteasomes. Number and activity of ENaC channel increases leading to increase sodium and water reabsorption in collecting duct.This results in volume expansion and hypertension. This action is independent of aldosteronism called pseudohypoaldosteronism. Volume expansion leads to suppression of rennin and aldosterone.
Clinical presentation: It presents early in life with hypertension, hypokalemia and alkalosis, although presentation in adulthood has been reported.
Lab finding: Hypokalemia metabolic alkalosis, low rennin and low aldosterone.
4. Syndrome of apparent mineralocorticoid excess
- Congenital: Loss of function mutations of 11-beta-hydroxysteroid dehydrogenase enzyme type 2 isoform (11-beta-HSD2), gene on chromosome 16q, autosomal recessive.
- Acquired disorder results from chronic exposure to glycyrrhetinic acid (e.g. from excessive consumption of black licorice) or carbenoxolone was used for peptic ulcer, which inhibits the activity of the 11-beta-HSD2 enzyme.
Pathophysiology: 11-beta-HSD2 normally converts cortisol to cortisone, a steroid that does not bind to the mineralocorticoid receptor. With 11-beta-hydroxysteroid type 2 enzyme deficiency or inhibition cortisol conversion to cortisone is decrease and cortisol levels are increased. Since cortisol levels are 1000 times greater than aldosterone, 11bHSD inhibition leads to mineralocorticoid receptor over-stimulation, causing hypokalemia, metabolic alkalosis and hypertension.
Clinical presentation: Rare disorder, childhood onset severe hypertension, extensive target organ damage, low birth weight, failure to thrive, polyuria due to diabetes insipidus induced by chronic hypokalemia. Other features are hypercalciuria, nephrocalcinosis and renal failure, hypertension, hypokalemia, metabolic alkalosis, and low plasma renin activity and low aldosterone.
Lab findings: Metabolic alkalosis, hypokalemia, decreased rennin and aldosterone level, hypercalciuria, high urinary free cortisol to free cortisone ratio in 24 hours urinary collection is sensitive test. In presence of enzyme deficiency urinary free cortisone is low or undetectable. Genetic testing confirms the diagnosis.
Microvascular compartment in kidney
Diseases affecting the microvacular compartment include:
- Atheroembolic disease
- Malignant hypertension
- Scleroderma renal crisis
- HUS/TTP (Thrombotic Microangiopathy) TMA
- Antiphospholipid antibody syndrome
- HELLP (Haemolysis, Elevated Liver Enzyme, Low Platelets)
- Vasculitis (presents mainly as glomerular involvement)
Clinical presentation: Acute kidney injury–acute renal failure
1. Atheroembolic renal disease
Atheroembolic is a complication of severe generalised atherosclerosis. Cholesterol crystal embolisation or atheroembolisation occurs when portions of an atherosclerotic plaque break off and embolise distally, resulting in partial or total occlusion of multiple small arteries leading to tissue or organ ischaemia. Renal involvement is common
Risk factors: Old age, hypertension, diabetes, smoking, atherosclerosis, atherosclerotic renal artery stenosis.
Clinical presentation: Can be acute or subacute, or chronic kidney injury.
Inciting events: Angiography, cardiovascular surgery, thrombolytic therapy, or anticoagulation or spontaneous event induced by haemodynamic stress. Cholesterol crystal embolisation is iatrogenic in more than 70 percent of cases. AKI occurs within one or two weeks after inciting events.
Clinical characteristics:
- New onset accelerated or labile hypertension, skin may be cyanotic, ulcerated digits, blue toes, livedo reticularis on back or lower extremities, multiorgan involvement, mesentery ischaemia, nausea, vomiting, gastrointestinal bleeding, pancreatitis, and/or central nervous system involvement producing transient ischaemic attack, confusion, visual symptoms, retina may show cholesterol emboli, oliguric to anuric renal failure
- The most common presentation of the disease is subacute kidney injury, in which progressive renal dysfunction occurs in staggered steps, separated by periods of stable kidney function (often referred to as a "staircase pattern")
- The least common presentation is the patient with chronic stable renal impairment and clinical features similar to those with ischaemic nephropathy (due to bilateral renal artery stenosis) and nephrosclerosis, which frequently coexist with cholesterol emboli
Laboratory findings: Urine- mild proteinuria (rarely nephrotic range), microscopic haematuria rarely RBC cast, eosinphiluria by Hansel stain, peripheral eosinophilia, raised CRP, hypocomplementemia, renal Doppler to rule out renal artery stenosis. Skin biopsy or kidney biopsy may be diagnostic. The cholesterol crystals within the emboli are dissolved during tissue fixation; what is therefore observed are path gnomonic biconvex, needle-shaped clefts ("ghosts") within the occluded vessel.
2. Thrombotic Microangiopathy (TMA)
Definition: Pathological hallmarks are thrombosis of arterioles and capillaries. Clinical hallmarks are mechanical haemolysis, microangiopathic haemolytic anemia, thrombocytopenia and acute renal injury or neurological manifestations. Multiple aetiologies associated TMA. Two most common conditions are haemolytic uraemic syndrome (HUS) and thrombotic thrombocytopenic purpura (TTP).
Current classification of TMA
- Primary thrombotic microangiopathy (TMA) syndromes
- Thrombotic thrombocytopenic purpura (TTP): Severe deficiency of ADAMTS13 (activity <10%)
- Classical pentad of fever, MAHA, thrombocytopenia, neurologic changes and acute kidney injury is rarely seen
- Inherited; may present in a newborn infant, a child with thrombocytopenia, or, less commonly, an adult
- Among adults, a common presentation is during a first pregnancy
- Inherited TTP has ADAMTS13 gene mutation
- Acquired autoimmune: Uncommon in children, seen mainly in adults; they have autoantibodies to ADAMTS13
- As compare to HUS TTP have severe thrombocytopenia, predominant neurological involvement, mild renal failure
- Complement-mediated TMA (atypical HUS)
- Inherited and acquired disorders may present in children or adults
- Inherited disorders usually have a heterozygous mutation in a gene encoding a regulatory protein in the alternate complement pathway (e.g. CFH, CFI, CD46/MCP, C3, CFB, CFHRs)
- Acquired is due to autoantibodies to complement factor H or I; this is the commonest type in India
- Renal failure is severe as compared to TTP
- Shiga toxin-mediated haemolytic uremic syndrome (ST-HUS) (typical diarrhoea positive D+)
- Associated with shiga toxin producing E.coli O157:H7 strain (90%)
- Abdominal pain; diarrhoea (often bloody); possible history of outbreak or exposure to livestock or contaminated food, although most cases are sporadic
- Renal failure is prominent
- Stool may be positive for the organism (Escherichia coli or Shigella dysenteriae) or shiga toxin
- Drug-induced TMA (DITMA)
- Immune mediated: Drugs implicated are Quinine, Gemcitabine, Quetiapine, Oxaliplatin. Immune-mediated TMA is typically severe. Symptoms suddenly begin, which commonly include chills, fever, abdominal pain, diarrhoea, nausea, and vomiting, and severe renal failure
- Dose-dependent, toxicity-mediated: Dose-dependent, toxicity-mediated DITMA syndromes due to direct cellular damage. These disorders can be acute, caused by a toxic dose of an approved or illegal drug, or chronic, occurring after weeks or months of drug administration. DITMA is primarily caused by chemotherapeutic agents (such as gemcitabine and mitomycin), immunosuppressive agents (such as cyclosporine and tacrolimus), vascular endothelial growth factor (VEGF) inhibitors (such as bevacizumab), and opioids taken inappropriately or illegal agents (such as oxymorphone and cocaine).
- Metabolism-mediated TMA: Inherited disorders of intracellular vitamin B12 (cobalamin) metabolism are exclusively hereditary due to mutations in the MMACHC gene (methyl malonic aciduria and homocystinuria type C). This leads to cobalamin deficiency. Patients may present with a TMA as an infant or adult. Elevated homocysteine and low methionine levels are seen in plasma, and urine may show methylmalonic aciduria.
- Coagulation-mediated TMA: Inherited, disorder involving mutations in genes encoding thrombomodulin (TM), plasminogen, and diacylglycerol kinase epsilon (DGKE) have been reported to be associated with TMA. Typically presents in children <1 year old. Thrombosis is seen in microvasculature and not in larger vessels like inherited thrombophilic disorder.
- Thrombotic thrombocytopenic purpura (TTP): Severe deficiency of ADAMTS13 (activity <10%)
- Secondary (Systemic diseases associated) TMA
- Systemic infection: May include bacterial, viral, rickettsial, or fungal organisms. High fever and shaking chills are common.
- Systemic malignancy: Breast, prostate, lung, pancreatic, or gastrointestinal tumors are often responsible.
- Pregnancy-related syndromes (e.g., severe preeclampsia, HELLP): Typically present in third trimester or postpartum. Severe hypertension and liver involvement are often present. Abnormalities resolve with delivery.
- Systemic rheumatic diseases (e.g. SLE, Scleroderma, APS): SLE may be associated with hypertension, renal insufficiency, and autoimmune cytopenias. APS typically presents with arterial and/or venous thromboembolism but can also produce a TMA.
- Solid organ transplant: May be associated with calcineurin inhibitor administration. May be associated with infection such as CMV in the setting of immunosupprression. In patients receiving a kidney transplant for a primary TMA syndrome, the syndrome may recur in the transplanted kidney.
Laboratory findings suggestive of TMA
- CBC: Anaemia, thrombocytopenia, raised LDH, reticulocytosis, fragmented RBCs, schistocytes in peripheral smear
- Renal chemistry: To asses severity of renal impairment: BUN, serum creatine, S Na schistocyes, K Cl.HCO3, serum heptoglobulin for degree of haemolysis, ADAMTS 13 activity
- Complement: C3, C4, CH50 antibodies against H, I mutations studies for complement factor H, F, I, B in selected patients
To read the previous article in this series, click here.
In the next part, Dr. NK Hase will write on lower urinary tract lesions, AKI, AKD, and CKD.
Disclaimer- The views and opinions expressed in this article are those of the author's and do not necessarily reflect the official policy or position of M3 India.
The author Dr. NK Hase is a Director clinical Nephrology & Transplant working at Jupiter Hospital, Thane and former Professor & Head of Department of Nephrology Seth GS Medical College and KEM Hospital, Mumbai.
-
Exclusive Write-ups & Webinars by KOLs
-
 Daily Quiz by specialty
Daily Quiz by specialty -
 Paid Market Research Surveys
Paid Market Research Surveys -
Case discussions, News & Journals' summaries
