Prion Disease: A Case Study on Evaluation & Management
M3 India Newsdesk Mar 21, 2024
Prion diseases, including sporadic Creutzfeldt-Jakob disease (sCJD), are rapidly progressive neurodegenerative conditions leading to early demise. This article provides insights into the clinical features, diagnostic challenges, and management strategies for sCJD.
Prion diseases refer to a group of neurodegenerative diseases in humans and animals. They are a common cause of rapidly progressive dementia and eventually lead to the early demise of the affected individual. The following table enlists the various types of prion diseases affecting both humans and animals.
Sporadic CJD is the most common variant affecting humans and shall be discussed in detail in this article.
Case
A 60-year-old gentleman, physically active, with no underlying co-morbid conditions, was noted by his wife to be mildly apathetic. He was speaking less than before and was losing interest in his previous daily activities.
Over the next couple of weeks, he started forgetting where he kept his objects of daily use, forgetting why he had entered a certain room in his house and was often seen roaming around his house purposelessly. One week later, he started having difficulty in reading words and sentences from the newspaper.
By the end of the first month, he had given up driving. He had started experiencing sudden jerky movements of his body (myoclonic jerks). Subsequently, he had speech difficulties, he spoke in short sentences and often mispronounced the words. He required assistance with bathing, dressing, and eating.
At the end of the third month, he started developing bouts of rage and got easily frustrated. His visual difficulties progressively worsened. His family history revealed that his mother had suffered from mild dementia for a span of 7 years.
Pathogenesis of prion disease
They are caused by the transformation of an endogenous protein (PrP, i.e. prion prion-related protein) into an abnormal configuration which is known as Prion. The abnormal protein PrPSc acts as a template for the misfolding of more amount of normal protein PrPC into PrPSc. PrPSc isoform is extremely resistant to proteolysis and degradation by conventional means of chemical and physical decontamination or disinfection.
Clinical features
sCJD is a rapid disease with a median survival of 5-6 months. Mostly individuals above the age of 60 are affected. When sCJD affects younger individuals, they have longer survival and present with non-cognitive features predominantly.
- The earliest symptom to appear in sCJD is cognitive decline. They include memory impairment, confusion, and difficulty in planning and organisation.
- Motor symptoms of sCJD include cerebellar ataxia, extrapyramidal symptoms (rigidity, dystonia, tremor, bradykinesia) and eventually myoclonus (sudden jerky movements of body parts).
- Behavioural or psychiatric symptoms may be seen (aggression, irritability, personality changes, depression, etc.).
- Constitutional symptoms are less obvious and include malaise, lightheadedness, fever, fatigue, etc.
- Some features of cortical affection may be seen, e.g. cortical blindness, aphasia, apraxia, neglect, etc.
- The end stage is represented by an akinetic-mute state (no purposeful movement and no speech).
Based on the predominant clinical feature, sCJD is classified into the following subtypes:
- Amaurotic or Heindenhain variant: rapidly progressive dementia, myoclonus, visual disturbances such as hallucinations, visual agnosia and cortical blindness.
- Brownell and Oppenheimer variant: early onset cerebellar ataxia and relatively late dementia.
- Thalamic variant: dementia and movement disorders.
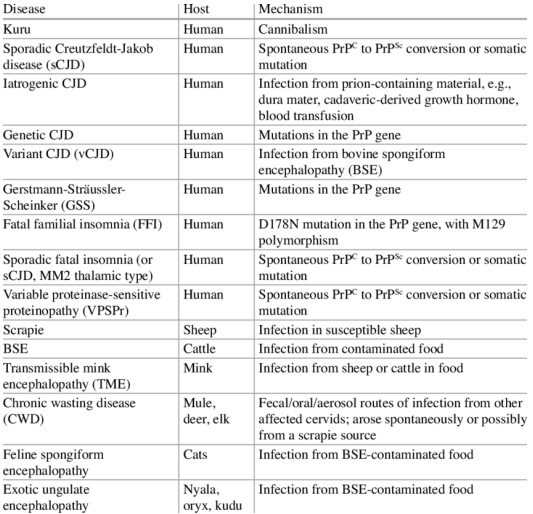
Another classification of sCJD is based on polymorphism at codon 129 of the prion (PRNP) gene and the type of protease-resistant prion. When PrPSc is extracted from the brain and partially digested with proteinase, depending on which of two sites (codon 82 or 97) at which PrPSc is cleaved, either a longer 21-kD (type 1) or a shorter 19-kD (type 2) peptide results when run on a Western blot.
On examination, the MMSE score was 15/30. He had deficits in the form of recent memory impairment, difficulty with orientation, object recall, calculations, naming, calculation, concentration and drawing the intersecting polygons. Besides he had mild nonfluent aphasia, strong bilateral grasp reflex, had more response to objects and threats in his left visual field and had mild dystonia of the right arm. His gait was slow with a normal base, occasionally requiring support.
Investigations & diagnosis
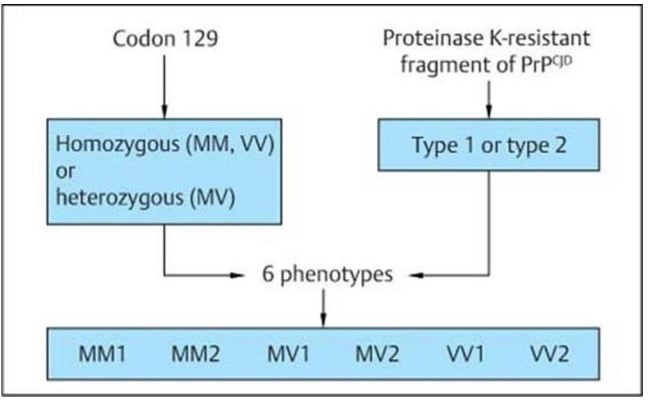
The pathology of sCJD includes neuronal loss, gliosis (proliferation of astrocytes), vacuolation (spongiform changes) and deposition of PrPSc. Non-specific features seen in other neurodegenerative diseases may be seen.
A, The periodic acid-Schiff (PAS)–stained section reveals multiple vacuoles, ranging in size from 5–20 µm, within the gray matter neuropil. Note the absence of PAS-positive amyloid plaques. B, Fine granular (synaptic) deposits and coarse deposits adjacent to some of the vacuoles are shown in this immunoperoxidase stain for protease-resistant prion protein (PrPSc).
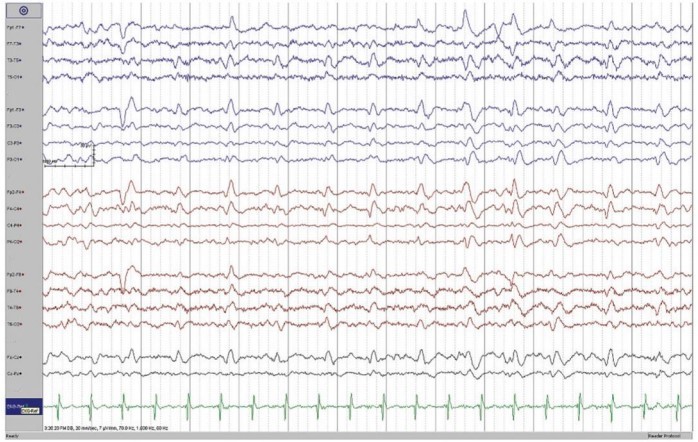
EEG reveals the presence of periodic sharp wave complexes (bi-phasic or triphasic) occurring at a frequency of about 1Hz.
CSF biomarkers for CJD include the 14-3-3 protein, total-tau (t-tau), neuronspecific enolase (NSE), and the astrocytic protein S100β. Their sensitivity and specificity have been shown to vary across all the studies conducted. Other biomarkers like CSF serum glial fibrillary acidic protein (GFAP) and serum neurofilament light chain (NfL) are under investigation and yet not in clinical use.
The ratio of total-tau and phosphorylated tau (T-tau/P-tau) is also being assessed as a diagnostic tool for CJD.
Real-time quaking-induced conversion (RT-QuIC) assay is a relatively newer technique by which the number of abnormal prion proteins is amplified and hence their detection is made easier. The substance to be tested is brought in contact with a substrate containing PrPC. This enables conversion into PrPSc which subsequently aggregates into amyloid fibrils and is detected with a fluorescent dye.
MRI imaging may reveal cortical gyral hyperintensities, more prominently seen in T2 and FLAIR sequences. This is described as cortical ribboning. Besides these, basal ganglia hyperintensities on T2-weighted sequences can also be seen in the early stages. The signal increase in CJD may also be seen in the hippocampus, thalamus, and mesencephalon.
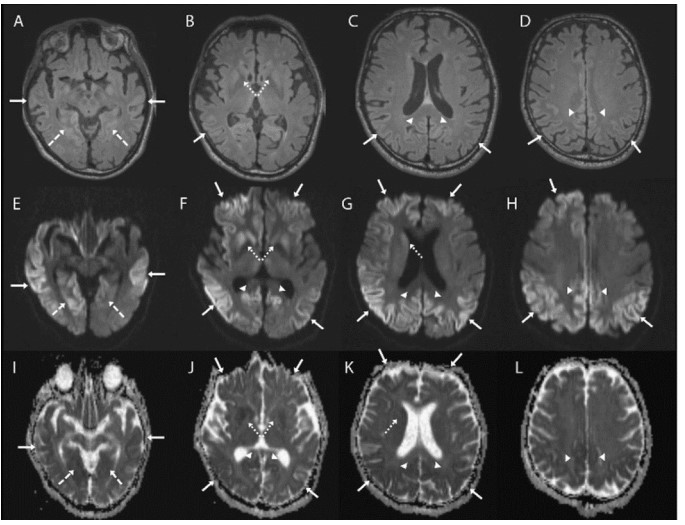
Axial brain MRI sequences in a patient with sporadic Jakob-Creutzfeldt disease. Note that the axial fluid-attenuated inversion recovery (FLAIR) sequences (A–D) are generally much less sensitive than the diffusion sequences, particularly diffusion-weighted imaging (DWI) (E–H) and the apparent diffusion coefficient (ADC) map (I–L). The DWI and ADC map show cortical ribboning (solid arrows) in the bilateral parietal, right greater than left frontal and lingula (dashed arrows), and posterior cingulate (arrowheads). Asymmetric involvement of the striatum is also shown (right greater than left) (dotted arrows). Bright regions on DWI are dark on the ADC map, indicating true restricted diffusion of water molecules.
Diagnostic criteria
Over the years, many diagnostic criteria have been developed to enable early diagnosis of sCJD. The most recent criteria were proposed in 2017 which categorises patients into definite, probable, and possible sCJD based on the level of diagnostic certainty.

Sporadic CJD diagnostic criteria (January 2017)
A brain MRI of the patient revealed a cortical-ribboning pattern with mild asymmetry and left caudate head hyperintensity. EEG showed intermittent sharp-wave complexes which were quasi-periodic along with slowing of background activity. Other routine blood biochemical tests were normal; lumbar puncture was not done as consent was not obtained.
The patient met the criteria for probable sporadic Creutzfeldt-Jakob disease. Symptomatic therapy was initiated. After 1 month, he succumbed to aspiration pneumonia. Brain autopsy and PRNP genetic testing confirmed the presence of sCJD.
Differential diagnosis
EEG features comprising periodic spike and wave complexes can also be seen in Hashimoto’s Encephalopathy, Lewy Body Disease and Hepatic Encephalopathy. Some other disorders which present with rapid cognitive decline like sCJD include paraneoplastic and autoimmune limbic encephalopathies, neoplasms, CNS vasculitis & metabolic and toxic encephalopathies. These must be thoroughly ruled out before considering a diagnosis of sCJD.
Management
Till date, there is no definite therapy for sCJD. Symptomatic therapy may temporarily benefit the patients. SSRIs (e.g. escitalopram) may be used to treat depression, anxiety, and mild agitation. Severe psychosis may be managed by atypical antipsychotics (e.g. quetiapine). Myoclonus is treated with valproic acid, levetiracetam or clonazepam.
Disclaimer- The views and opinions expressed in this article are those of the author and do not necessarily reflect the official policy or position of M3 India.
About the author of this article: Dr. Annesh Bhattacharjee is a Consultant Neurologist at Cosmo Medical, Guwahati.
-
Exclusive Write-ups & Webinars by KOLs
-
 Daily Quiz by specialty
Daily Quiz by specialty -
 Paid Market Research Surveys
Paid Market Research Surveys -
Case discussions, News & Journals' summaries
